2ヶ月前に、VCFを扱うにあたってbcftools便利ですよという記事を書いたのですが、その中でも最近よく使うquery機能についてご紹介します。
前の記事では
「多様な使い方があるので、簡単には説明できません」
なんて雑にくくってしまいましたのでもうちょっと丁寧にします。
まずはをusageを出してみましょう。
$ bcftools query
About: Extracts fields from VCF/BCF file and prints them in user-defined format
Usage: bcftools query [options] <A.vcf.gz> [<B.vcf.gz> [...]]
Options:
-e, --exclude <expr> exclude sites for which the expression is true (see man page for details)
-f, --format <string> see man page for details
-H, --print-header print header
-i, --include <expr> select sites for which the expression is true (see man page for details)
-l, --list-samples print the list of samples and exit
-o, --output-file <file> output file name [stdout]
-r, --regions <region> restrict to comma-separated list of regions
-R, --regions-file <file> restrict to regions listed in a file
-s, --samples <list> list of samples to include
-S, --samples-file <file> file of samples to include
-t, --targets <region> similar to -r but streams rather than index-jumps
-T, --targets-file <file> similar to -R but streams rather than index-jumps
-u, --allow-undef-tags print "." for undefined tags
-v, --vcf-list <file> process multiple VCFs listed in the file
Examples:
bcftools query -f '%CHROM\t%POS\t%REF\t%ALT[\t%SAMPLE=%GT]\n' file.vcf.gz
VCFまたはBCFから、任意の形式で列を抽出して出力する、だそうです。
一番下にexamplesのコマンドがあるので、試してみましょう。
こんな感じのVCF(test.vcf)でやってみます。
##fileformat=VCFv4.2 略 ##contig=<ID=chr1,length=248956422,assembly=genome.fa> ##contig=<ID=chr2,length=242193529,assembly=genome.fa> ##contig=<ID=chr3,length=198295559,assembly=genome.fa> ##contig=<ID=chr4,length=190214555,assembly=genome.fa> ##contig=<ID=chr5,length=181538259,assembly=genome.fa> ##contig=<ID=chr6,length=170805979,assembly=genome.fa> ##contig=<ID=chr7,length=159345973,assembly=genome.fa> ##contig=<ID=chr8,length=145138636,assembly=genome.fa> ##contig=<ID=chr9,length=138394717,assembly=genome.fa> ##contig=<ID=chr10,length=133797422,assembly=genome.fa> ##contig=<ID=chr11,length=135086622,assembly=genome.fa> ##contig=<ID=chr12,length=133275309,assembly=genome.fa> ##contig=<ID=chr13,length=114364328,assembly=genome.fa> ##contig=<ID=chr14,length=107043718,assembly=genome.fa> ##contig=<ID=chr15,length=101991189,assembly=genome.fa> ##contig=<ID=chr16,length=90338345,assembly=genome.fa> ##contig=<ID=chr17,length=83257441,assembly=genome.fa> ##contig=<ID=chr18,length=80373285,assembly=genome.fa> ##contig=<ID=chr19,length=58617616,assembly=genome.fa> ##contig=<ID=chr20,length=64444167,assembly=genome.fa> ##contig=<ID=chr21,length=46709983,assembly=genome.fa> ##contig=<ID=chr22,length=50818468,assembly=genome.fa> ##contig=<ID=chrX,length=156040895,assembly=genome.fa> ##contig=<ID=chrY,length=57227415,assembly=genome.fa> ##contig=<ID=chrM,length=16569,assembly=genome.fa> ##reference=file:///home/genome/hg38/genome.fa ##source=HaplotypeCaller #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SRR077861 chr21 10392066 . G A 54.81 LowCoverage AC=1;AF=0.500;AN=2;BaseQRankSum=-0.319;ClippingRankSum=0.000;DP=4;ExcessHet=3.0103;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=27.00;MQRankSum=0.000;QD=13.70;ReadPosRankSum=1.150;SOR=1.609 GT:AD:DP:GQ:PL 0/1:1,3:4:20:83,0,20 chr21 10392138 . A G 19.81 LowCoverage;VeryLowQual AC=1;AF=0.500;AN=2;BaseQRankSum=0.524;ClippingRankSum=0.000;DP=5;ExcessHet=3.0103;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=27.00;MQRankSum=0.000;QD=3.96;ReadPosRankSum=-0.674;SOR=0.446 GT:AD:DP:GQ:PL 0/1:3,2:5:48:48,0,80 chr21 10392275 . G A 211.77 PASS AC=1;AF=0.500;AN=2;BaseQRankSum=1.181;ClippingRankSum=0.000;DP=15;ExcessHet=3.0103;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=26.64;MQRankSum=0.842;QD=14.12;ReadPosRankSum=0.069;SOR=1.179 GT:AD:DP:GQ:PL 0/1:6,9:15:99:240,0,139 chr21 10392571 . A G 96.77 PASS AC=1;AF=0.500;AN=2;BaseQRankSum=-0.494;ClippingRankSum=0.000;DP=11;ExcessHet=3.0103;FS=3.424;MLEAC=1;MLEAF=0.500;MQ=29.83;MQRankSum=-0.605;QD=8.80;ReadPosRankSum=1.254;SOR=2.636 GT:AD:DP:GQ:PL 0/1:6,5:11:99:125,0,169 chr21 10393089 . G T 10.20 LowQD;VeryLowQual AC=1;AF=0.500;AN=2;BaseQRankSum=1.731;ClippingRankSum=0.000;DP=13;ExcessHet=3.0103;FS=3.736;MLEAC=1;MLEAF=0.500;MQ=29.41;MQRankSum=-0.213;QD=0.78;ReadPosRankSum=1.089;SOR=0.086 GT:AD:DP:GQ:PL 0/1:11,2:13:38:38,0,397
SRR077861というサンプルの、染色体21番上の変異が5個記載されたVCFです。
それでは、いざ。
$ bcftools query -f '%CHROM\t%POS\t%REF\t%ALT[\t%SAMPLE=%GT]\n' SRR077861_6.vcf chr21 10392066 G A SRR077861=0/1 chr21 10392138 A G SRR077861=0/1 chr21 10392275 G A SRR077861=0/1 chr21 10392571 A G SRR077861=0/1 chr21 10393089 G T SRR077861=0/1
すっきりしたタブ区切りテキストが出てきました。
解説しましょう。
bcftools queryの-fオプションは、出力形式を自由に指定することができるオプションです。
-fの後の' 'に囲まれた部分で、VCFをどんな形で出力するか指定しています。
上記のコマンドでは '%CHROM\t%POS\t%REF\t%ALT[\t%SAMPLE=%GT]\n' ですね。
- たくさん出てきている
\tと末尾の\nは、プログラミングの経験がある方ならご存じ、この2文字でそれぞれタブと改行を表します。 - タブ(
\t)で区切られている%CHROM%POS... は、(VCFを見たことがある人ならぴんと来たと思いますが)VCFの列名です。出力したい列名に%を付けることで、VCFの各行の、指定した列を出力することを指示しています。- サンプルごとに異なる情報が記載されるFORMAT以降の列は
[]で囲って表します。 つまりこういうことです。
- サンプルごとに異なる情報が記載されるFORMAT以降の列は
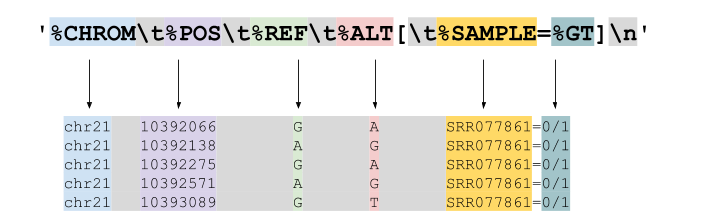
- ちなみに
%をつけ忘れると「VCFの列名」ではなく「ただの文字列」と認識されます。私はよくやらかすので、皆様もお気をつけてください。
$ bcftools query -f 'CHROM\tPOS\tREF\tALT[\tSAMPLE=GT]\n' SRR077861_6.vcf CHROM POS REF ALT SAMPLE=GT CHROM POS REF ALT SAMPLE=GT CHROM POS REF ALT SAMPLE=GT CHROM POS REF ALT SAMPLE=GT CHROM POS REF ALT SAMPLE=GT
- 当然、
-fの指定は、上記コマンド例以外にも応用が利きます。- 「なんか全体的にdepth薄い気がするんだよなーVCF全体のdepthの分布を知りたいなー」というときは
$ bcftools query -f '[%DP]\n'などいかがでしょうか。 - 個人的にちょっと便利だなと思った出力形式は
%TYPEです。TYPEなんて列名、VCFで見たことないと思いますが、何を表しているのかは、よろしければご自分で確かめてみてください。
- 「なんか全体的にdepth薄い気がするんだよなーVCF全体のdepthの分布を知りたいなー」というときは
bcftools queryの公式のドキュメントはこちら。
アメリエフでは、バイオデータ解析やそのシステム・インフラ環境の開発に興味のあるエンジニア・リーダー候補を募集しています。
www.wantedly.com