こんにちは、システム開発チームの adachit です。
第2回テーマは「Nextflowの基本的な使い方」です。
Nextflowを利用するための準備として、インストールやログの見方、設定などを解説します。
Nextflowのインストール
Nextflowを使用するためには、前提条件としてJavaが必要となります。
Macの場合、パッケージマネージャのHomebrewを使い、brew install openjdkでJavaをインストールします。
続けてbrew install nextflowでNextflowをインストールし、nextflow -vでバージョンを確認します。
Windowsの場合、Javaは公式ホームページからダウンロードしてインストールできます。
その後、コマンドプロンプトやPowerShellを使い、curl -fsSL get.nextflow.io | bashでNextflowをインストールします。
Linux(特にUbuntu)の場合、sudo apt install default-jreでJavaをインストールし、次にcurl -fsSL get.nextflow.io | bashでNextflowをインストールするのが一般的です。
また、解析プラットフォームとしてDockerを使用する場合は、事前にインストールしておく必要があります。
Macの場合、「docker docs」にアクセスし、dmgファイルをダウンロードしてインストールします。
ただし、ライセンスには注意が必要です。
アカデミアの場合は無料ですが、一部の企業では有料となります。
詳しくは公式サイトをご参照ください。
Windowsの場合、Mac版と同様に、exeファイルをダウンロードしてインストールします。
Linuxの場合、詳細は割愛しますが、sudo apt installコマンドで簡単にインストールできます。
ワークフローのインストール
Nextflowをインストールした後、例えば「rnaseq」ワークフローを使いたい場合は、nextflow run nf-core/rnaseqを実行します。
これにより必要なソフトウェアが自動的にダウンロードされワークフローが実行されます。
なお、既にNextflowを使用している場合は、nextflow listでダウンロード済みのパイプライン一覧を確認してください。
もし「rnaseq」が既に存在する場合は、nexflow pull nf-core/rnaseqで最新版に更新してください。
pullで最新版に更新後、runでワークフローを実行することを推奨します。
Nextflowの基本的な使い方
次に、Nextflowのログに関して解説します。
公式サイトで提供されている「tutorial.nf」を実行すると、ログが出力されます。
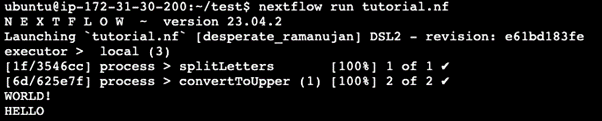
nextflow run tutorial.nfを実行すると、最初にNextflowのバージョン(例:23.04.2)が表示されます。
5-6行目では、どのソフトウェアが実行されたか、例えばsplitLettersやconvertToUpperが実行されたことが分かります。
また、「timeline.html」というファイルが生成されるので、ブラウザで開けば各プロセスの実行時間や使用したメモリなどの詳細を確認できます。
さらに、Nextflowを実行するディレクトリに「nextflow.config」というファイルを作成することで、リソースの管理やパラメータの設定が可能です。
例えば、CPUを4コア、メモリを16GBと指定したり、パイプラインに渡すパラメータのファイルのパスの指定、ツールのパスの設定などができます。
カスタマイズの方法は多岐にわたりますが、詳細は公式のマニュアルを参照してください。
次回予告
第3回テーマは「実践:RNA-seqパイプラインをNextflowで実行」です。実際にNextflowでの解析を、順を追って解説します。
さらに、第4回はパイプラインのカスタマイズを解説したいと思いますので、楽しみにしていてください。
※本記事は、2023年8月2日開催の第88回バイオインフォマティクス勉強会「Nextflowで始めるNGSデータ解析ワークフロー」講演内容をベースに作成しております。
動画で本記事の内容を視聴したい、講演資料PDFをダウンロードしたい方は、アメリエフの運営する会員制動画サイト「バイオインフォマティクス実践ラボ」にご登録ください。
