こんにちは、システム開発チームの adachit です。
第3回テーマは「実践:RNA-seqパイプラインをNextflowで実行」です。
実際にNextflowでRNA-seq解析をする流れを、順を追って解説します。
RNA-seqパイプラインの概要
さっそく、「rnaseq」のパイプラインをNextflowで実行してみましょう。
「nf-core」の「rnaseq」の詳細ページに図を見ると、パイプラインの概要を理解しやすいと思います。
このブログにそのまま図を貼り付ける訳にもいかないので、説明用に以下のように描きなおしました。
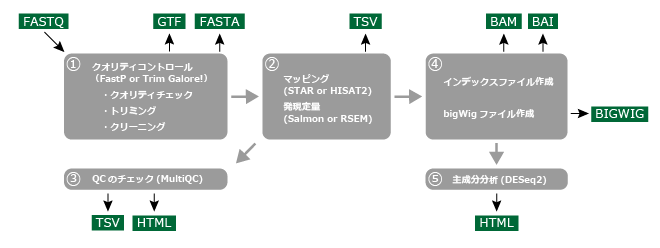
「rnaseq」パイプラインは5つのブロックに分かれていて、それぞれにDockerコンテナが配置されています。
1番のブロックでは、入力したFASTQファイルに対しクオリティコントロール(FastP or Trim Galore!)を実行します。
これにより、FASTQファイルのクオリティチェックやトリミング、クリーニング作業が行われます。
2番のブロックでは、リードデータのマッピング(STAR or HISAT2)および発現定量(Salmon or RSEM)、
3番のブロックでは、MultiQCを使ったクオリティチェック、
4番のブロックでは、マッピング後のデータファイルに対し、インデックスファイルの作成やbigWigファイルの作成などを行い、
5番のブロックでは、DESeq2による主成分分析までが行われます。
このパイプラインでは、いわゆる二次解析までの作業がカバーされています。
発現マトリクスは得られますが、三次解析にあたるヒートマップなどの描画、二群間比較解析などは含まれていませんので、研究の実務ニーズに合わせて利用・拡張することが重要です。
RNA-seqパイプラインの実行手順
実際の解析の手順を追ってみましょう。
まず、FASTQファイルを準備します。
次に、指定された書式のCSVファイルを作成します。
このCSVには1列目にサンプル名、2・3列目にFASTQファイルへのパス(forward / reverse)、そして4列目にストランド情報を記入します。
そして、nextflow run nf-core/rnaseqコマンドを実行します。
実行時には、先ほど作成したCSVファイル、出力フォルダ、ゲノムのバージョン、そして解析を実行するプラットフォーム(例: Docker)を指定します。
出力フォルダは自動で作成され、必要なリファレンスやDockerイメージも自動でダウンロードされて解析が開始されます。
解析が完了すると、ログが出力されます。
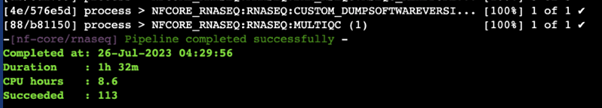
「Pipeline completed successfully」と表示されていれば、解析が問題なく完了したことがわかります。
ログの「Duration」項目を確認すると、解析にかかった時間が表示されます。
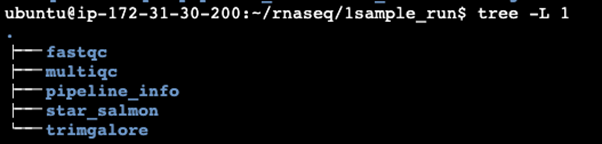
最後に、各解析に応じたフォルダが作成されますので、treeコマンドを使って実行結果を確認します。
次回予告
第4回テーマは「実践:Nextflowのパイプラインをカスタマイズする」です。
Nextflowの長所「自分でパイプラインをカスタマイズする」実例を挙げながら解説したいと思いますので、楽しみにしていてください。
※本記事は、2023年8月2日開催の第88回バイオインフォマティクス勉強会「Nextflowで始めるNGSデータ解析ワークフロー」講演内容をベースに作成しております。
動画で本記事の内容を視聴したい、講演資料PDFをダウンロードしたい方は、アメリエフの運営する会員制動画サイト「バイオインフォマティクス実践ラボ」にご登録ください。
