Trinityは de novo transcriptomeアセンブリング(RNA-seqデータからリファレンス配列のない生物遺伝子を構築する)ツールです。
以前のブログでインストール方法を紹介しています。
アセンブリを行ったあとは、
作成された配列を新たなリファレンス配列として、RNA-seqデータの(マッピング・)発現定量・サンプルごとの発現比較解析を行います。
アセンブリに使うのと発現定量〜 に使うデータは同じものです。
論文の解析ワークフロー図がわかりやすいです。↓
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3875132/figure/F2/
マッピング・発現定量 と 発現比較解析 は、他のバイオインフォマティクスソフトウェアを使って行うわけですが、
実はTrinity のコマンドを経由して他のソフトウェアを実行させることができます。(パスを通しておく必要があります)
中間データファイルの変換がスムーズであることや、Rコマンドを覚えなくても実行できることがメリットです。
以下はコマンドの一例ですが、Trinityのコマンドを使って 発現定量ソフト「RSEM」とマッピングソフト「bowtie2」を動作させています。
$TRINITY_HOME/util/align_and_estimate_abundance.pl \ --transcripts 2_trinity/Trinity.fasta \ --seqtype fq \ --sample_file samplen.txt \ --est_method RSEM --aln_method bowtie2 \ --output_dir outputdir \ --trinity_mode --prep_reference
このように利用できるのは以下のソフトウェアです。
マッピング
- bowtie
- boetie2
発現定量
- RSEM
- eXpress
マッピングをせずに発現定量
- kallisto
- Salmon
発現比較
- edgeR
- limma.voom
- DESeq2
- ROTS
遺伝子発現変動度合いを表す図も簡単に作成できます。
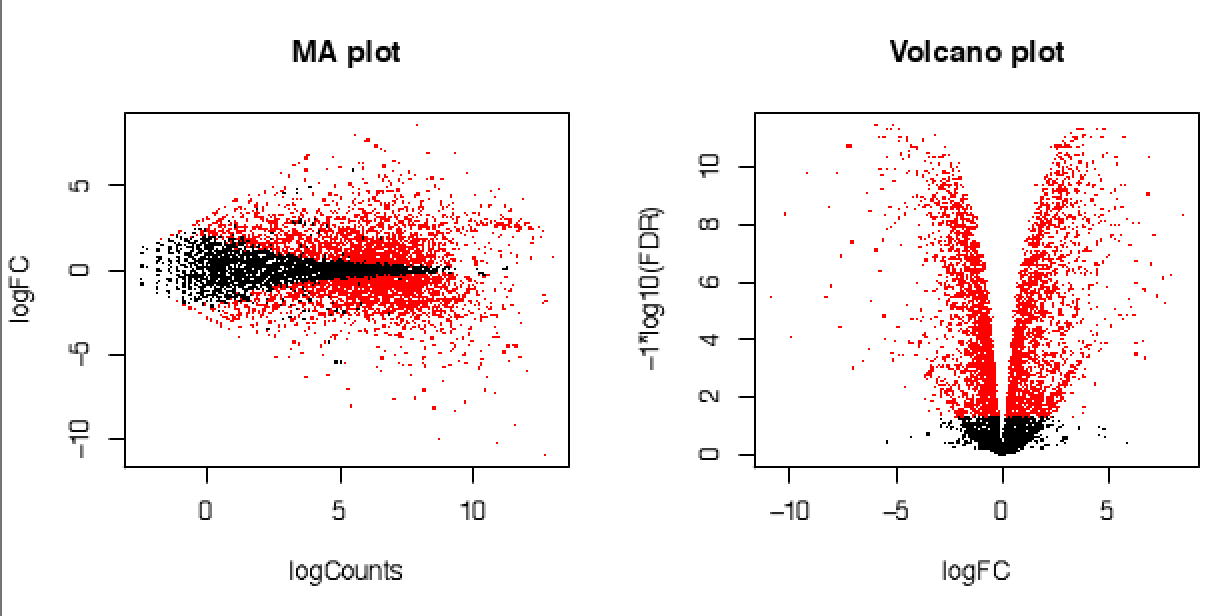
便利ですね。
お問い合わせは会社HPまで。
RNA-seqの発展版としてトレーニング実施実績もあります。