RNA-seq のバイオインフォマティクスデータ解析で2群比較の場合、比較の順番が重要になります。
遺伝子Cの発現がこの棒グラフのようにあった場合、
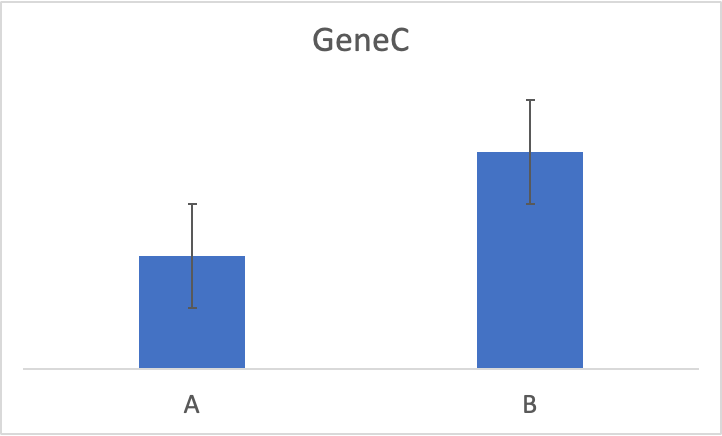
- AはBに比べ遺伝子Cが0.5倍 (グループA vs グループB )
- BはAに比べ遺伝子Cが2倍 (グループB vs グループA )
上記は、同じことではありますが、調べたい群を「コントロールに比べ 何倍」と言うことで解釈がしやすくなりますね。
一般的には「<刺激後> は <刺激前=コントロール> に比べて何倍」という順番で比較します。
edgeR
edgeR を用いたRNA-seq の発現比較においては、Rオブジェクトの作成時に比較群を定義します。
・想定データ:発現マトリクス
| Geneid | ... | Sample_A1 | Sample_A2 | Sample_B1 | Sample_B2 |
|---|---|---|---|---|---|
| GeneC | ... | 100 | 120 | 199 | 201 |
| GeneD | ... | 200 | 200 | 200 | 200 |
これをRに読み込みます。
library(edgeR) data <- read.delim("発現マトリクスファイル") #適宜header , row.names オプションなど指定する
次にサンプルのグループを指定するため、長さがサンプル数と同じ、因子型(factor)を含むベクトルを作成します。
ポイントとして、edgeR ではLevelsが若い方がコントロール群として認識されます。
levels=オプションを指定しない場合、Levelsは自動で辞書順になります。 *1
グループBがコントロール群だとすると次の様になります。
> treatment <- factor(c("A","A","B", "B"), levels=c("B", "A"))` > treatment [1] A A B B Levels: B A
- factor の第一引数:各サンプルのグループ名のベクトル。順番は発現マトリクスにおけるサンプルの並び順と同じ
- levels オプション:コントロール群が先、比較群が後のベクトル
treatment の中身を確認すると、サンプルのグループ名順があっているのはもちろん、Levels: で B → A の順番になっています。
このあとは、DGEList関数で発現マトリクスを 解析用Rオブジェクトに変換していきますが、ここでtreatmentを引数に指定します。
発現比較までの流れは次の様になります。
y <-DGEList(counts=data[,サンプルが含まれる列番号], group=treatment) cpm <- cpm(y) keep <- rowSums(cpm > 1) >= 2 y_expressed <- y[keep, ] #低発現遺伝子の除外 y_norm <- calcNormFactors(y_expressed) d_com <- estimateCommonDisp(y_norm) d_mod <- estimateTagwiseDisp(d_com) out <- exactTest(d_mod) final <- topTags(out, n=nrow(out$table))
解析結果を確認すると、
A-B (AがBに比べてどれだけ変動した)の比較になっていること、
GeneCは log2FCがマイナス、すなわちAはBに比べて発現が低いことが確認できます。
> head(final) Comparison of groups: A-B logFC logCPM PValue FDR GeneC -1.2720 5.2175 4.5777e-11 8.0315e-7 ...
※ 遺伝子名・数字は適当です。
参考 Bioconductor - edgeR 公式ドキュメント
*1:なので、難しいことは考えずコントロール群 = 1, 比較群 = 2 というグループ名で指定する!という形で覚えても良いです